行业动态
活动计划可能会使痴呆症患者望而却步
佐治亚州州立大学老年学研究人员领导的一项研究显示,辅助性生活社区可以通过与个体接触并尝试将所有居民纳入活动范围,来改善痴呆症患者的
佐治亚州州立大学老年学研究人员领导的一项研究显示,辅助性生活社区可以通过与个体接触并尝试将所有居民纳入活动范围,来改善痴呆症患者的

来自瓦伦西亚政治大学(UPV)和CIBER生物工程,生物材料与纳米医学(CIBER-BBN)的团队已在临床前设计和测试了一种用于治疗和恢复肌肉损伤的新
根据新的研究,增加的黄烷醇(一种天然存在于水果和蔬菜中的分子)的摄入量可以保护人们免受精神压力诱发的心血管事件的影响,例如中风,心脏
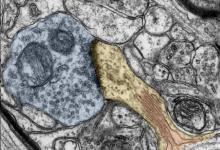
神经科学家一致认为,一个人的大脑在不断变化,重新布线并适应环境刺激。这是人类学习新事物并创造回忆的方式。这种适应性和延展性称为可塑
糖几乎在杂货店的货架上尖叫,特别是那些销售给孩子的产品。儿童是添加糖的最高消费者,即使高糖饮食与肥胖,心脏病,甚至记忆功能受损等健

科罗拉多州丹佛市,2020年3月31日-与典型的同龄人相比,唐氏综合症患儿患急性淋巴细胞白血病(ALL)的可能性高20倍,患急性髓系白血病(AML)的
科罗拉多大学医学院研究人员的一个团队最近发表了一篇论文,对缺氧或缺氧在癌症发展中的作用提供了新的见解。CU癌症中心的成员Joaquin Esp
科罗拉多州奥罗拉(2021年3月30日-科罗拉多大学护理学院的研究人员发现,近四分之一的护理学研究生报告说,在过去的一年中,COVID-A加剧了压
利用阳光驱动水净化的一项新发明可以帮助解决从电网提供清洁水的问题。该设备类似于一块吸水的大海绵,但留下了铅,油和病原体等污染物。为
在本月初发表在《美国牙科协会杂志》上的一项纵向研究中,研究人员分析了参加ForsythKids预防牙科计划的近7,000名儿童的未经治疗的蛀牙。在
高胆固醇是最常见的动脉粥样硬化原因,动脉硬化会增加心脏病和中风的风险。但是现在,圣路易斯华盛顿大学医学院的科学家已经发现了一个基因
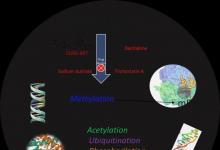
酗酒严重影响人类健康。酒精中毒,更好地定义为酒精使用障碍(AUD),包括一组与酒精引起的损害有关的病理实体。患有AUD的人会在停止饮酒时表
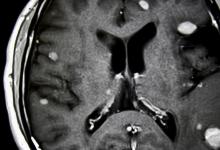
科罗拉多州奥罗拉(2021年3月31日)-一项在《精神病学前沿》上发表的新研究发现,强迫症(OCD)以及其他精神病合并症(例如自闭症谱系或抽动障碍
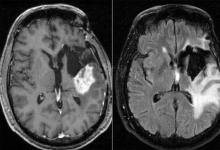
胶质母细胞瘤是一种侵袭性的脑癌,无法治愈。患者在诊断后平均存活15个月,少于10%的患者存活时间超过5年。虽然研究人员正在通过正在进行的

新的汗贴可以使科学家们轻松地收集和分析婴幼儿皮肤上的汗水,从而简化对囊性纤维化的早期诊断。当与51位受试者进行测试时,该贴纸与以前的